Az NMR-spektroszkópia alkalmazási lehetôségei a gyógyszertervezésben
Edward M. Purcell1 és Felix Bloch2 vezette
azokat a történeImi jelentôségû kísérleteket
1946-ban, ameIyek a mágneses magrezonancia (NMR) spektroszkópia
megszületéséhez vezettek. Az NMR-spektroszkópia
alig több mint ötvenéves múltja alatt bekövetkezett
bámulatos fejlôdés egyetlen spektroszkópiai
technikával sem állítható párhuzamba.
Az elmúlt ötven év bebizonyította, hogy az NMR-spektroszkópia
a molekulák szerkezetének és a molekuláris
kölcsönhatások vizsgálatának rendkívül
hatékony és sokoldalúan alkalmazható kutatási
eszköze. Folyamatosan bôvülô fizikai, kémiai,
biológiai és orvosi alkatmazásaival az NMR-technika
napjainkra önálló, multidiszciplináris tudománnyá
vált: számtalan szakkönyv és tudományos
folyóirat foglalkozik az NMR elméletével és
gyakorlati alkalmazásaival; legatább nyolc fizikai és
két kémiai Nobel-díjat adományoztak olyan kutatóknak,
akik munkája különbözô mértékben
kapcsolódik a mágneses magrezonancia jelenségéhez.3
Az NMR-spektroszkópia már hosszú ideje a gyógyszeripari
kutatások egyik kiemelten fontos eszköze. Történelmi
okokból fakadóan elsôdleges szerepe a kémiailag
szintetizált kis szerves vegyületek vagy különféle
természetes forrásokból izolált molekulák,
metabolitok azonosítása és szerkezetének jellemzése.
Az NMR hagyományos gyógyszeripari alkalmazása elsôsorban
a szintetikus kémiai kutatómunkát, valamint a szennyezésprofil-vizsgálatokat
és a metabolitkutatást támogatja. Az elmúlt
néhány évtizedben végbemenõ forradalmi,
technológiai és módszertani fejlôdésnek
köszönhetôen az NMR-spektroszkópia gyógyszeripari
alkalmazási köre jelentôsen kibôvült. Egyre
több olyan külföldi gyógyszeripari példáról
olvashatunk, ahol az NMR-spektroszkópiát más gyógyszertervezési
eszközökkeI karöltve sikeresen alkalmazzák egy racionálisabb
gyógyszerfejlesztés folyamatában. Az NMR-spektroszkópiával
meghatározható szerkezeti, dinamikai és egyéb
kémiai-biológiai szempontból hasznos információk
a szerkezet-hatás összefüggés megértését
elôsegítve, jelentôs hatást gyakorolhatnak a
gyógyszerkutatás-fejlesztés irányára.
A jelen összefoglaló tanulmány kitekintést kívánt
nyújtani azokra az NMR-spektroszkópiában rejlô
potenciális lehetôségekre és módszerekre,
melyek elsôsorban gyógyszertervezési szempontból
a hazai gyógyszerkutatásban meghonosításra
érdemesek. A jelen közlemény alapjául külföldi
összefogIaló tanulmányok szolgáltak.4
***
NMR a gyógyszerkutatásban
A 20. század végét jellemzõ, egyre gyorsuló tudományos és technikai fejlõdésnek köszönhetõen a gyógyszeripari kutatás és fejlesztés (K+F) alapvetõ stratégiai változásokon ment keresztül. A multinacionális gyógyszervállalatok átalakult K+F stratégiája egyértelmûen kijelöli a 21. század gyógyszerkutatásának irányát. Ezt az utat olyan technológiák kifejlõdése és térhódítása fémjelzi, mint például a kombinatorikus kémia, a nagy áteresztõképességû in vitro biológiai tesztelés (HTS, High-Throughput Screening), és a racionális gyógyszertervezés. Az új technológiák és kutatási stratégiák meghonosítására, anyagi kereteik között, a hazai gyógyszergyárak is törekednek, aminek sikeres példáját mutatja a Chinoin prolil endopeptidáz enzim inhibitorok fejlesztése a racionális gyógyszertervezés eszköztárának felhasználásával.5
Ahhoz, hogy az NMR nyújtotta lehetõségeket el tudjuk helyezni az új típusú gyógyszerkutatási stratégiákban, vázlatosan át kell tekintenünk a gyógyszerfejlesztési folyamatot. Az 1. ábra a korszerû gyógyszerfejlesztés-tervezés ciklikus folyamatának egy nagyon leegyszerûsített sémáját, valamint azokat a kapcsolódási pontokat mutatja, ahol az NMR potenciálisan hasznos információkat képes szolgáltatni.
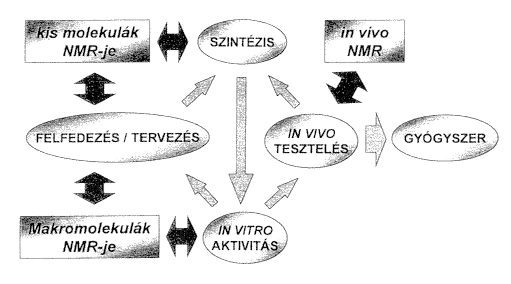
1. ábra. Az NMR-spektroszkópia kapcsolódása a gyógyszerfejlesztés folyamatához
A korszerû gyógyszerkutatás felfedezõ szakaszában a "minél többet, minél gyorsabban" elvét alkalmazzuk: lehetõleg kombinatorikus kémiai szintézisekkel létrehozott vegyülettárakból in vitro HTS-teszteken kiválasztjuk azokat a célzott biológiai hatású vezérmolekulákat, amelyek a gyógyszerfejlesztés kiindulópontjául szolgálnak. A gyógyszerkutatás következõ tervezési fázisában a "minél jobbat, minél okosabban" elve érvényesül. A szerkezet-hatás összefüggés felderítésén keresztül, számítógépes molekulatervezés segítségével, a vezérvegyületek szerkezeti, fizikai-kémiai paramétereinek finom változtatásával, optimalizálásával jutunk el egy olyan ígéretes gyógyszerjelöltig, amely már részletes in vivo tesztelésre kerülhet, és gyógyszerré fejieszthetô. A kombinatorikus kémia és a HTS lehetõvé teszik a gyógyszerfejlesztés kezdeti szakaszának felgyorsítását, míg a számítógépes molekulatervezés eszközei a szerkezet-hatás összefüggés megértését mozdítják elô. Az 1. ábra azt illusztrálja, hogy az NMR a gyógyszerfejlesztés kezdeti felfedezési, és azt követô tervezési stádiumában, valamint az in vivo tesztelés fázisában is képes integráltan részt venni, és hasznos információkkal szolgálni.
A gyógyszerfejlesztési folyamat szempontjából alapvetôen az NMR-kísérletek három csoportját lehet megkülönböztetni:
a) A kis molekulák NMR-je általában a gyógyszeriparban tipikusan elôforduló kisebb molekulatömegû (1 kDa ³ molekulatömeg) szerves vegyületek szerkezetvizsgálatát jelenti. Ez tehát az NMR hagyományos gyógyszeripari alkalmazási területe, mely elsõsorban a szintetikus kémiai munkát támogatja a K+F különbözô stádiumaiban, de egyre nagyobb szerepet kap a minôségbiztosításban is.
b) A makromolekuláris NMR lehetõvé teszi biopolimerek, elsõsorban fehérjék és nukleinsavak, illetve ezek egymással és kisebb szerves és szervetlen vegyületekkel alkotott komplexeinek vizsgálatát. A makromolekuláris NMR-rel meghatározható információk a felfedezési stádiumban az in vitro tesztelést, míg a tervezési fázisban a szerkezet-hatás összefüggés megértését mozdítják elô.
c) Az in vivo NMR lehetõséget teremt a gyógyszerjelölt hatásának in vivo vizsgálatára szöveteken, célszerveken, élô állatokon és emberen, ami a metabolitkutatásban, illetve a gyógyszerfejlesztés végsõ klinikai stádiumaiban szolgál hasznos információkkal.
Az összefoglaló tanulmány fókuszpontjában a gyógyszertervezési szempontból legfontosabb makromolekuláris NMR módszerei állnak. Ez a terület a hazai NMR-mûszerparkkal és mûszertechnikai hátérrel elérhetõ, illetve a jövõben egyre inkább elérhetôvé válik. Az NMR hagyományos, analitikai jellegû gyógyszeripari alkalmazásait (lásd molekulák szerkezetvizsgálata) nem tárgyaljuk. A rendkívül dinamikusan fejlõdõ in vivo NMR gyógyszerfejlesztési vonatkozásait csak röviden érintjük.
Receptoralapú gyógyszertervezés
A makromolekuláris NMR-rel meghatározható információk
hatékonyan alkalmazhatók a gyógyszerkutatási
stratégiákban egyre fontosabb szerephez jutó ún.
szerkezet-, vagy receptoralapú gyógyszertervezésben.
A receptoralapú gyógyszertervezés kiindulópontja a biológiai célmolekula (fehérje, nukleinsav) azonosítása, elôállítása és tisztítása szerkezetkutatási célokra. Amennyiben a biológiai célmolekulát sikerül NMR-spektroszkópiai vagy röntgenkrisztallográfiai vizsgálatokra alkalmas formában elõállítani, és térszerkezetét meghatározni, úgy lehetõvé válik a vezérmolekulák szerkezet-, vagy receptoralapú optimalizálása. Ennek során meghatározzuk a preparatív biokémiai tesztek alapján kiválasztott vezérmolekula receptorral alkotott komplexének térszerkezetét. A receptor és a ligandum bioaktív konformációjának ismeretében, a molekuláris felismerésért felelôs kölcsönhatások megértésével lehetõség nyílik a ligandum szerkezetalapú optimalizálására, olyan analógok tervezésére, amelyek várhatóan nagyobb affinitásúak. Az optimatizált ligandumot szintetikusan elõállítjuk, majd biológiai tesztelés után a ciklust addig folytatjuk, míg további affinitásnövekedést már nem tudunk elérni.
A makromolekuláris NMR a receptoralapú gyógyszertervezés folyamatába három ponton képes aktívan bekapcsolódni:
egyik kapcsolódási pont a szabad biológiai célmolekula
térszerkezetének meghatározása,
a másik pont a receptor-ligandum komplex szerkezeti és
egyéb vizsgálata,
a harmadik kapcsolódási pont a vezérmolekulák
azonosítása.
A továbbiakban a biológiai szempontból talán
legfontosabb célmolekulák, a fehérjék, illetve
fehérje-ligandum komplexek vizsgálatára fogunk szorítkozni.
Makromolekuláris NMR
Mindenekelôtt tekintsük át azokat az erõforrásokat, amelyek megléte a makromolekuláris NMR mûvelésének alapfeltétele (2. ábra).
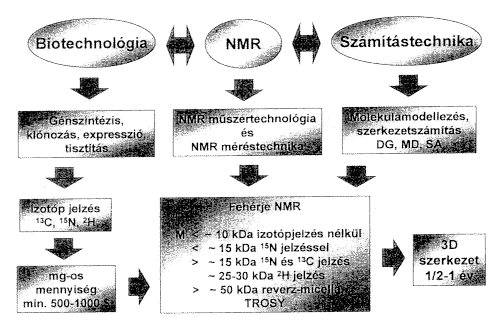
2. ábra. Fehérjék NMR-rel történô meghatározásának feltételei
A makromolekulásis (fehérje) NMR alapvetôen
három terület szimbiózisán alapszik:
a) Az egyik terület a biotechnotógia, ami lehetõvé
teszi fehérjék nagy mennyiségû elôállítását
(génszintézis, mutáció, klónozás,
expresszió), tisztítását, szelektív,
vagy teljes izotópjelzését (13C, 15N,
2H)
NMR szerkezetkutatási célokra.
b) Az NMR-spektroszkópia, amely képes makromolekulák
szerkezeti és dinamikai paramétereinek meghatározására.
(A fehérjék térszerkezetének NMR-rel történô
meghatározása nagyszámú, atomok közötti
távolságok és torziós szögek meghatározásán
alapszik. Ezeket az atomi távolság és torziós
szög adatokat a fehérjeszerkezet számításakor
mint a fehérje szerkezetét definiáló ún.
kényszerfeltételeket használjuk.)
c) Harmadrészt a számítástechnika, ezen
belül olyan spektrális adatfeldolgozási és molekulamodellezési
módszerek, amelyek lehetõvé teszik az NMR-rel kísérletileg
meghatározott spektrumokból molekulaszerkezeti információk
meghatározását, illetve ezeket felhaszálva,
a makromolekula, illetve a makromolekula-ligandum komplex térszerkezetének
atomi szintû leírását, számítását
(szerkezetszámítás).
A három technológia együttesen képes arra,
hogy oldatfázisban a fehérjék háromdimenziós
térszerkezetét meghatározza kb. 10 kDa molekulatömegig
iztópjelzés nélkül, 1015 kDa között
minimum 15N izotópjelzést alkalmazva, 15 kDa fölött
13C
és 15N izotópjelzéssel (2530 kDa környékén
már deutériumjelzés is szükséges). Az
elmúlt egy-két évben kezdenek kifejlôdni azok
a technikák, amelyek a proteinek szerkezetmeghatározásának
felsô hátárát kb. 100 kDa molekulatömegig
képesek felvinni.6 Napjainkra az NMR-spektroszkópia
a röntgenkrisztallográfiával összemérhetõ
felbontással képes a biopolimerek térszerkezetének
atomi szintû leírására. (A makromolekulák
szilárd fázisban történõ vizsgálatának
metodikai fejlesztése jelentôs erõvel folyik, és
nagy fejlõdés elôtt áll különösképpen
a membránproteinek tekintetében.)
A makromolekuláris NMR-rel kapcsolatban meg kell említeni gyógyszeripari szempontból alapvetõen fontos két tényezôt: a vizsgálatok költség- és idõvonzatát. A fehérjék géntechnológiai úton történô elôállítása fejiett biotechnológiai hátteret igényel. A szerkezetkutatásra alkalmas fehérje elõállítása, a megfelelõ expressziós rendszer kifejlesztése és az izotópjelzés költséges mûveletek. Egy NMR-mérésre alkalmas, izotópjelzéssel ellátott minta elõállítási költségét 1000 USD-ben mérjük. Ezt a mintát egy dedikált kutatócsoport kimondottan erre a célra fenntartott készüléken, melynek értéke felszereltségtôl függõen több 100 mFt, több hónapig méri folyamatosan. A szerkezetvizsgálat NMR-es alapfeltétele a korszerû NMR-es mûszertechnika és méréstechnika. Ez minimum 500 MHz-es többcsatornás készülék, a megfelelõ izotópok (1H, 2H, 13C, 15N) mérésére alkalmas mérôfejek, gradiens egység, hullámforma generátor stb. meglétét, másrészt naprakész, az adott egyedi készülékre adaptált méréstechnikát, speciális pulzusszekvenciák implementálását jelenti. Ezen erôforrások megléte esetén ideális esetben a vizsgált fehérje térszerkezetét, a fehérje méretétôl, tulajdonságaitól függõen szerencsés esetben fél, de inkább minimum egy év alatt lehet meghatározni. A szerkezetszámításhoz szintén több tízmillió Ft nagyságrendû szakértõi szoftverre és hardverre (adatfeldolgozás + molekulamodellezés, munkaállomás) van szükség. Ezek az adatok és tények látszólag nehezen egyeztethetôek össze a gyógyszeripari kutatást meghatározó idôbeli és financiális határokkal. Mégis a jelentôsebb külföldi gyógyszervállalatok saját makromolekuláris NMR-apparátust mûködtetnek. A makromolekuláris NMR ugyanis nem csak néhány akadémiai intézetben mûvelt alapkutatási tevékenység, hanem napjainkban már bevett protokollok alapján mûködõ, "rutin" eljárássá vált, amely, mint technológia know-how, a jelentõsebb külfoldi gyógyszergyárak K+F tevékenységének szerves részét képezi.
Egyre inkább érvényesül az a tendencia, hogy a gyógyszeripari kutatások ismert szerkezetû biológiai célmolekulákhoz köthetô hatásmechanizmusokat céloznak meg. Nyilvánvalóan egy új, addig ismeretlen biológiai célmolekula izolálása és szerkezetmeghatározása, a szerkezet-hatás összefüggés és hatásmechanizmus megállapítása jelentôs elônyökkel jár az originális gyógyszerkutatásban. A makromolekuláris NMR-laboratóriumok azonban csak részben foglalkoznak új biólógiai célmolekulák szerkezetmeghatározásával. Gyógyszerkutatási szempontból legalább ennyire, vagy még inkább fontos a már akadémiai kutatásból, vagy más egyéb forrásból ismert térszerkezetû biopolimerekhez történõ ligandumkötõdés vizsgálata. Protein- és NMR-adatbankokban számos receptor szerkezeti és NMR-paraméterei hozzáférhetôek, mely a ligandumkötódés-vizsgálatok kiindulási pontjául szolgálhatnak.
Hazai viszonylatban ezeknek az erõforrásoknak egy része
rendelkezésre áll. Például a Richter Gedeon
Rt.-ben üzemelô Varian gyártmányú, 500
MHz-es NMR-mûszer már jelenlegi felszereltsége mellett
is képes izotóp által jelzett fehérjék
mérésére, továbbá az NMR-mérések
kiértékelését, és a szerkezetszámítást
lehetõvé tévõ mindkét vezetô molekulamodellezô
és NMR-spektrumértékelõ szoftvercsomag (MSI,
TRIPOS) megtalálható. Az irodalomból ismert fehérjék
expressziójára kifejlesztett törzsek megvásárolhatóak,
új törzsek kifejlesztése pedig célszerûen
külsõ kutatás keretében finanszírozható
lehet. Így gyakorlatilag csak fermentációra és
fehérje tisztítására van szükség,
ami már gyógyszergyári apparátussal is megoldható.
Makromolekuláris NMR-információk
... A kötôdés tényének igazolására bármilyen NMR-rel mérhetõ molekuláris paraméter, pl. kémiai eltolódás, jelszélesség, T1 és T2 relaxációs idõ, nukleáris Overhauser-effektus (NOE), diffúziós állandó stb. megváltozása szolgálhat. Amikor a kis molekulatömegû ligandum hozzákötõdik a nagy mólsúlyú makromolekulához, akkor azok a fizikai paraméterek fogják jellemezni, mint a makromolekulát, ezért a jelszélesség megnõ, a T2 relaxációs idõ lecsökken, a NOE-effektus elõjelet vált (+ > ), a diffúziós állandó lecsökken. Attól függôen, hogy a ligandum kis affinitással (Kd > 105), vagy nagy affinitással (Kd < 108) kötõdik a fehérjéhez, eltérõ képet kapunk az NMR-spektrumban, ezért eltérõ stratégiát kell alkalmazni a kötõdés vizsgálatánál. Kis affinitású ligandumok esetében a szabad és kötött forma (az NMR-mérés ideje alatt) gyorsan egymásba alakul, így az NMR-spektrumban a két állapot átlagát mutató jelet detektálunk, míg nagy affinitás esetében a két forma jelei elkülönülnek.
Kötôdésvizsgálatok lehetõvé teszik
a makromolekula-ligandum komplex disszociációs egyensúlyi
állandójának
és a disszociáció sebességének meghatározását,
amibôl a kötôdés erôsségére,
a makromolekula-ligandum komplex stabilitására következtethetünk.
Ezek a kvantitatív információk felhasználhatóak
pl. QSAR (Quantitative Structure-Activity Relationship) számításoknál.
A disszociációs állandó meghatározására
protokollok léteznek, amelyek szerint a ligandum koncentrációjának
függvényében felvett titrálási görbék
analízisével határozzák meg a Kd-értéket,
legáltalánosabban a kémiai eltolódás
és jelszélesség változását figyelve.7
A kombinatorikus kémia és HTS technológiájával kapcsolatban felmerülõ nagyon izgalmas és rendkívül aktuális kérdés a vegyülettárak, keverékek "dekonvolúciója". A gyószerkutatás felfedezõ szakaszában a "minél többet, minél gyorsabban" elve szerint a vezérmolekulák kiválasztására egy HTS-teszten egyszerre több komponens keverékét is vizsgálhatjuk (pl. kombinatorikus kémiai keverék szintézissel létrehozott vegyülettárat). Amennyiben a HTS-teszt pozitív választ ad, alapvetõ kérdést jelent, hogyan határozzuk meg, hogy a vizsgált keverék melyik komponense felelõs a hatásért. Elõfordulhat, hogy a HTS-en kapott pozitív válasz tulajdonképpen fals pozitív, ami a komponenskeverék egészétôl származik. A keveréket alkotó komponenseket külön-külön vizsgálva az aktivitás elvész.
Több NMR-es technika is létezik, amely lehetõvé teszi keverékekbôl az aktív komponens kiszûrését, szerkezetének azonosítását anélkül, hogy a keveréket fizikailag komponenseire kellene szétválasztanunk, ami jelentôs idômegtakarítást jelent. Ezek a módszerek kis affinitású ligandumok esetében alkalmazhatóak, tehát kimondottan a vezérvegyületk azonosítására alkalmasak. Az affinitás alapján történõ szûrésre a relaxációs idôk és a diffúziós állandó, és a NOE elôjelének megváltozását használják.8 Az Abott cég például eljárás-szabadalommal védi az általuk kifejlesztett NMR-es technikát.9 Ezek a módszerek a feleslegben lévô ligandumok jeleit, illetve azok változását mérik, tehát nem igényelnek köItséges izotópjelzett fehérjét.
Alapvetõen fontos kérdés a kötõdés helyének meghatározása, hiszen erre a HTS általában nem ad választ. Az NMR viszonylag gyors és hatékony módszert nyújt ennek a kérdésnek a megválaszolására. A fehérjék amid jeleinek kémiai eltolódása nagyon érzékeny indikátora a kötõdésnek. A ligandumkötôdés hatására a kötõdés lokális környezetében lévô amid jelek kémiai eltolódása megváltozik, ami a fehérje szerkezetének és a spektrum asszignációjának ismeretében közvetlen információt ad a kötõdés helyérôl. Ennek az effektusnak a mérésére szolgál egy igen érzékeny és gyors mérés, az ún. kétdimenziós 1H-15N HSQC (Heteronuclear Single Quantum Coherence) kísérlet, amely 15N-jelzett fehérjék amid jeleinek vizsgálatát teszi lehetõvé. A receptor kötõhelyének feltérképezésére ugyancsak felhasználható az amid hidrogén-deutérium kicserélôdés sebességének megváltozása.
A másik izgalmas témakör a térszerkezet vizsgálata,
ami egyrészt a makromolekula és ligandum bioaktív
konformációjának meghatározását,
a kötõdésért felelõs funkciós csoportok
azonosítását, az egyes csoportok ionizációs
állapotának vizsgálatát, valamint a vegyület
oldószerrel érintkezõ felületének meghatározását
jelenti.
A szerkezet-hatás összefüggés analízise
során ezek az információk rendkívüli értéket
képviselnek, és nagymértékben elõsegíthetik
a molekuláris hasonlóságon alapuló analógok
megtervezését.
A ligandum bioaktiv konformációját, kis affinitású ligandumnál ún. NOE transzfer vizsgálatokkal állapíthatjuk meg,10 míg nagy affinitású ligandumok esetében ún. izotóp szerkesztett és izotóp szûrt méréseket11 használhatunk a ligandum és receptor jeleinek megkülönböztetésére és a konformáció vizsgálatára.
A fehérje és ligandum egyes funkciós csoportjainak ionizációs állapota fontos információ lehet az enzimmechanizmus és a molekuláris felismerés megértésében. A pKa-érték, a tautomer formák meghatározása jó példája annak, hogyan egészítheti ki és bõvítheti az NMR a pusztán geometriai információkat.
Gyógyszertervezési szempontból hasznos megkülönböztetni a vezérmolekula oldószerrel érintkezô felületét, mely vélhetôen kevésbé fontos szerepet játszik a receptor felismerésben, és azokat a régiókat, amelyek a receptor affinitásért és specificitásért felelõsek. Az oldószerrel érintkezõ felületeken olyan kémiai módosításokat hajthatunk végre, amelyek a gyógyszerjelölt oldhatóságát, biológiai hasznosíthatóságát, farmakokinetikáját és toxicitási tulajdonságait javíthatják. Az oldószerrel érintkezô, illetve kötésben részt vevô felületek megállapítására relaxációs reagenseket (pl. HyTEMPO) használhatunk.
A harmadik témakör a dinamikai vizsgálatok. Ez az
a pont, ahol az NMR a röntgenkrisztallográfia pusztán
szerkezeti információján alapvetôen túlmutat.
Az oldatfázisban megvalósuló különbözõ
szintû molekuláris mozgások felderítése
fontos szerepet játszhat a molekuláris felismerésért
felelõs folyamatok megértésében. Ilyen mozgások
például a proteinek gerinckonformációjának
vagy oldalláncok konformációjának dinamikája.
Ligandumok esetében többszörös kötôdési
módok meghatározása, annak dinamikája válik
vizsgálhatóvá. Ezeknek a molekuláris mozgásoknak
a megváltozása a kötôdés hatására
fontos szerkezet-hatás információval bír. Ugyancsak
vizsgálható a fehérjék hidratálódása,
a fehérje harmadlagos szerkezetét stabilizáló
ún. szerkezeti víz dinamikai sajátságai.12
SAR by NMR módszer
Végezetül külön említését érdemel egy rendkívül ígéretes gyógyszertervezési stratégia, amelyet az Abbott cég fejlesztett ki és szabadalmaztatott.13 ...
A szerkezet-hatás összefüggés NMR-re alapozott
vizsgálata elegáns módon ötvözi a racionális
tervezés és a kombinatorikus kémia elemeit. A már
említett 1H-15N HSQC-kísérlet
segítségével, az amid jelek kötôdés
hatására történô megváltozását
figyelve vizsgálnak egyszerre 10-20 kis molekulából
álló keveréket. Ezzel a módszerrel, mérésidõt
tekintve, egy nap kb. 1000 molekula tesztelhetõ. Miután azonosították,
hogy a keverék melyik komponense kötõdik a fehérjéhez,
meghatározzák Kd értékét.
Majd, hogy affinitásnövekedést érjenek el, a
vezérmolekulához hasonló szerkezetû analógokat
vizsgálnak. Ezután az optimalizált elsõ ligandum
jelenlétében további ligandumokat keresnek NMR-rel,
amelyek az elsõ ligandum kötôdési helye közelében
kötôdnek. A második ligandumot is optimalizálják,
majd a két ún. vezérfragmens kiválasztása
után, röntgenkrisztallográfia vagy NMR-spektroszkópia
segítségével meghatározzák a vezérfragmensek
helyét és orientációját a fehérje-ligandum
harmadlagos komplexben. A két vezérfragmens egymáshoz
és a fehérjéhez viszonyított orientációjának
ismeretében olyan molekulát terveznek, amelyben a két
vezérfragmenst megfelelõ kémiai lánc köti
össze. Az így kapott molekula kötõdést tekintve
bifunkcióssá válik, ami jelentõs affinitásnövekedést
eredményez. Például a szervtranszplantációk
során az immunválasz elnyomásában fontos szerepet
játszó FKBP (FK506 kötõ protein) protein esetében
egy Kd = 2 mM és egy Kd
= 0,1 mM affinitású fragmens összekötésével
Kd = 19 nM affinitást tudtak elérni.14
Hasonlóképpen, Stromelysin esetében egy Kd
= 17 mM és egy Kd = 0,02 mM affmitású fragmensbõl
egy Kd = 15 nM affinitású vegyületet terveztek
kevesebb, mint hat hónap alatt.15
In vivo NMR
Az in vivo NMR-nek két fõ irányvonala rajzolódik ki. Az egyik irányvonal rövidítése MRI (Magnetic Resonance Imaging). Az MRI olyan noninvazív képalkotó eljárás, ami forradalmasította a klinikai diagnosztikát és a patológiát. Az MRI alkalmas élô szervezetek tetszôleges irányú metszetérõl rétegfelvételt készíteni, mely a legkülönbözõbb morfológiai jellegû elváltozások (infarktus, daganat, fejlôdési rendellenesség stb.) kimutatására használható. Gyógyszerkutatási szempontból az MRI különféle betegségek vizsgálatában, diagnosztikájában alkalmazható állatmodelleken vagy emberen, elõsegítve új gyógyszervegyületek hatékonyságának klinikai jellemzését.
A másik irányvonal neve MRS (Magnetic Resonance Spectroscopy),
ami egy adott szövet vagy szerv lokalizált térfogatelemében
képes molekuláris információt, vagyis spektrumot
nyújtani (természetesen kisebb felbontással, mint
a folyadékfázisú, nagyfelbontású NMR-készülékek).
Az MRS gyógyszerkutatási alkalmazása leginkább
a metabolitkutatásban (metabolitok alatt tágabb értelemben
a szervezet endogén anyagait értve, pl. ATP, ADP, kreatin-foszfát,
N-acetil-szpartát stb.), azon keresztül a betegségek
patomechanizmusának molekuláris szintû felderítésében
nyilvánul meg.
Összefoglalás
A szakirodalomban fellelhetõ adatok tanúbizonysága
szerint az NMR-spektsoszkópia folyamatosan bôvülõ
eszköztárával a modern gyógyszerkutatási
stratégiák egyik fontos pillérét képezi.
Az NMR alkalmazási köre kiterjed a gyógyszerfejlesztés
korai (vezérmolekulák azonosítása és
optimalizálása) és késõi (metabolizmus,
klinikai jellemzés) szakaszára is. A molekuláris felismerésért
felelõs receptor-ligandum kölcsönhatások atomi
szintû vizsgálatán keresztül kiemelkedôen
fontos információkat nyújt a szerkezet-hatás
összefüggés felderítéséhez és
az új terápiás készítmények kifiejlesztésében
egyre sikeresebben alkalmazott szerkezet-, vagy receptoralapú gyógyszertervezéshez.
A hazai gyógyszerkutatás tekintetében a makromolekuláris
NMR-spektroszkópia meghonosítása és integrálása
a kombinatorikus kémia, a HTS és a racionális gyógyszertervezés
eszközeivel, a külföldi példák szerint, célszerûnek
látszik. A rendelkezésre álló hazai erõforrások
alapján a makromolekuláris NMR meghonosítása
leginkább (legkisebb költséggel, leggyorsabban) kötôdésvizsgálatokban
(pl. vezérmolekulák azonosítása) kezdôdhet
eI.
HIVATKOZÁSOK
1. E. M. Purcell, H. G. Torrey, R. V. Pound, Phys. Rev., 69, 37 ( 1946).
2. a) F. Bloch, W. Hansen, M. E. Packard, Phys. Rev., 69, 127 (1946);
b) F. Bloch, Phys. Rev., 70, 460 (1946).
3. I. I.. Rabi (1944, fizika); E. M. Purcell (1952, fizika), F. Bloch
(1952, fizika), A. Kastler (1966, fizika), J. H. Van Vleck (1977, fizika),
N. Bloembergen (1981, fizika), K. A. Müller (1987, fizika), N. R Ramsey
(1989, fizika), H. G. Dehmelt (1989, fizikaj, R. R. Ernst (1991, kémia),
J. Pople ( 1998, kémia).
4. a) NMR in drug design, Ed. D. J. Craik, CRC Press, New York (1996);
b) NMR methods for elucidating macromolecule-ligand interactions: an approach
to drug design, Eds. R. E. Handschumacher, I. M. Armitage, Proceedings
of the fourth biochemical pharmacology symposium, New Haven, CT, July (1989);
c) B. J. Stockman: NMR spectroscopy as a tool for structure-based drug
design, Prog. NMR Spectr., 33. 109-151 (1998); d) M. Billeter, NMR for
structural studies in drug discovery, 3. 151-167 (1995); e) S. W. Fesik,
NMR structure-based drug design, J. Biomol. NMR, 3. 261-269 (1993); f)
S. W. Fesik, NMR studies of molecular complexes as a tool in drug design,
J. Med. Chem., 34. 2937-2945 (1991).
5. Hermecz István, Kánai Károly, Arányi
Péter, Gyógyszerkutatás a Chinoinban, Magyar Tudomány,
1998. 9. sz.
6. a) K. Pervushin, R. Ruek, G. Wider, K. Wütrich, Proc. Natl.
Acad. Sci. USA, 94, 12366-12371 (1997); b) K. Pervushin, R. Ruek, G. Wider,
K Wütrich, J. Am. Chem. Soc., 120, 6394-6400 (1998).
7. NMR of Macromolecules, Chapter 6, The Practical Approach Series,
ed. G. C. K. . Roberts, Oxford University Press (1993).
8. a) H. Ponstingl, G. Otting, J. Biomol. NMR, 9, 441-444 (1997); b)
P. J. Hajduk, E. T. Olejniczak, S. W. Fesik, J. Am. Chem. Soc., 119, 12257-12261
(1997); c) M. Lin, J. Shapiro, J. R. Wareing, J. Am. Chem. Soc., 119, 5249-5250
(1997); d) B. Meyer, T. Weimar, T. Peters, Eur. J. Biochem, 246, 705-709
(1997); e) N. Gonnella, M. Lin, M. J. Shapiro, J. R. Wareing, X. Zhang,
J. Magn. Reson., 131, 336-338 (1998).
9. S. W. Fesik, P. J. Hajduk, E. Olejniczak WO 98/48264.
10. a) G. M. Clore, A. M. Gronenborn, J. Magn. Reson., 48, 402-417
(1982); b) A. M. Gronenborn, G. M. Clore, Biochem. Pharmacol., 40(1), 115-119,
(1990).
11. a) G. Ottireg, H. Senn, G. Wagner, K. Wüthrich, J. Magn. Reson.,
70, 500-515 ( 1986); b) S. W. Fesik E. R. P. Zuiderweg E. T. Olejniczak
R. T. Gampe, Biochem. Pharmacol. 40(1) 161-168, (1990).
12. G. Wider, Progress in NMR Spectroscopy, 32, 193-275 ( 1998).
13. a) S. W. Fesik, P. J. Hajduk, E. Olejniczak WO 97/18469; b) P.
J. Hajduk, R. P. Meadows, S. W. Fesik, Science, 278, 497-499 (1997 October);
c) S. B. Shuker, P. J. Hajduk, R. P. Meadows, S. W. FesiK, Science, 274,
1531-1534 (1996 November)
14. S. B. Shuker P. J. Hajduk, R. P. Meadows, S. W. Fesik, Science,
274, 1531-1534 (1996 November).
15. a) P. J. Hajduk et. al, J. Am. Chem. Soc., 119, 5818-5827 (1997);
b) E. T. Olejniczak et. al., J. Am. Chem. Soc., 119, 5828-5832 (1997).