Bajza István
Mycobacteriális oligoszacharidok elõállítása
Kossuth Lajos Tudományegyetem, Debrecen
1996
Ezen dolgozat Mycobacteriális eredetû sejtfelszíni antigének oligoglikozil hapténjeinek elõállításával foglalkozik.
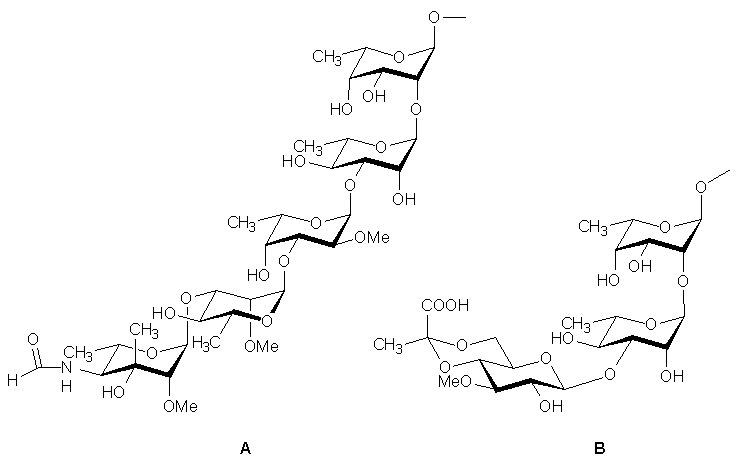
Az elsõ fejezet a Mycobacterium avium 14-es szero-variáns glikopeptidolipid típusú antigén oligoszacharid komponesének szintézisét tárgyalja. Ez egy pentaszacharid (A ábra), melyben egy különleges szerkezetû egység, az N-formilkansosamin és a természetben ritka D-ramnopiranóz is szerepel. Az N-formilkansosamin elõállításakor két fõ problémát kellett megoldanunk: az egyik a láncelágazás, a másik a 4-dezoxi-4-formamido funkció kialakítása volt. A C-metilezést Klemer C-alkilezési eljárásával végeztük el, mellyel jó hozammal sikerült elõállítani a metil-6-dezoxi-2,3-O-izo-propilidén-3-C-metil-a-L-lyxo-hexopiranozid-4-ulózt.
A másik problémát az ekvatoriális 4-dezoxi-4-form-amido funkció kialakítása jelentette. Sikertelen volt az a próbálkozásunk, melyben megkíséreltük a metil-6-dezoxi-2,3-O-izopropilidén-3-C-metil-a-L-talopiranozidot Mitsunobu reakcióval átalakítani a megfelelõ aziddá. A megoldást egy nem túl elegáns, de célravezetõ módszer jelentette: a metil-6-dezoxi-2,3-O-izopropilidén-3-C-metil-a-L-lyxo-hexopira-nozid-4-ulóz oxim LiAlH4-del végzett redukciójakor ugyanis nem tapasztaltunk szelektivitást és mind a 4,6-didezoxi-manno-, mind a 4,6-didezoxi-talo- konfigurációjú amin képzõdött, mely elegybõl célvegyületünk 41 %-os hozammal volt izolálható. Néhány egyszerû védõcsoport manipuláció után az irodalomból jól ismert módon alakítottuk ki a 3-O-benzil-4-N-benzilformamido-4,6-didezoxi-3-C-metil-2-O-metil-a-L-mannopiranóz-triklór-acetimidát glikozil donort.
A fentiekkel teljesen analóg módon elõállítottuk a fenil-1-tio-kansosamin donort is.
A p-nitrofenil-4-O-benzil-6-dezoxi-2-O-metil-a-D-man-nopiranozidot p-nitrofenil-a-D-mannopiranozidból állítottuk elõ. Itt a megoldandó problémát a könnyen redukálható p-nitrofenil aglikon melletti 6-dezoxi funkció kialakítása jelentette a megfelelõ 6-O-tozil származékból Ennek redukciójakor sérült az aglikon is, ezért I-ra cseréltük a OTs-csoportot, melyet sikeresen redukáltunk, majd szelektív metilezés után nyertük a megfelelõ glikozil akceptort.
A terminális 3-O-benzil-4-N-benzilformamido-4,6-didez-oxi-3-C-metil-2-O-metil-a-L-mannopiranozil-(1®3)-4-O-benzil-6-dezoxi-2-O-metil-a-D-mannopiranozil-(1®OPNP) és a pentaszacharid 1 + 4 szintézisekor a fenil-1-tio-kan-sosaminil donorral végzett glikozilezések nem vezettek eredményre. Az N-formilkansosaminil imidátot használva donorként 35 % -os hozammal sikerült izolálni a fenti diszacharidot, de sikertelen volt az 1 + 4 kapcsolás. Ezért elõállítottuk a 3-O-benzil-4-N-benzilformamido-4,6-didez-oxi-3-C-metil-2-O-metil-a-L-mannopiranozil-(1®3)-4-O-benzil-6-dezoxi-2-O-metil-a-D-mannopiranozil-(1®SEt) di-szacharid donort is és 2 + 3 blokkszintézissel sikeresen szintetizáltuk célvegyületünket, az immundetermináns pentaszacharidot.
A második fejezet a Mycobacterium avium 8-as szero-variáns GPL triszacharid komponensének (B. ábra) szintézisét írja le. A terminális 3-O-metil-glükopiranozil egység 4,6-O-piruvát acetálját a tanszéken kidolgozott módszer módosításával állítottuk elõ, reagensként metil-piruvát-dietil-ditioacetálját alkalmazva. Elõször modellreakciókat végeztünk az új reagenssel, modellvegyületnek a metil-2,3-di-O-benzil-a-D-glükopiranozidot alkalmaztuk.
Metil-triflátot használva tiofil ágensként nem volt teljes a konverzió nagy mennyiségû (5 ekv./SEt) metil-triflát alkalmazásakor sem. Az IDCP-s és a NIS/TfOH aktiválási mód eredményre vezetett, a reakciókban az S és R diasztereomerek aránya 1 : 1 volt. A SO2Cl2/TfOH rendszert alkalmazva 1 aktiválására az S : R arány a számunkra kedvezõ 2 : 1 volt, ezért ez utóbbit alkalmaztuk az oligoszacharid komponensének szintézisére.
A p-Nitrofenil-endo-3,4-O-benzilidén-6-dezoxi-2-O-(2,4-di-O-benzil-a-L-ramnopiranozil)-a-L-talopiranozid diszacharid szintézisekor új ramnozil donort (Etil-3-O-acetil-2,4-di-O-benzil-1-tio-a-L-ramnopiranozid) alkalmaztunk, módosítva az irodalmi szintézist. A 3-O-metil-4,6-O-piruvil-D-glükopiranozil egység aktiválására a triklór-acetimidátos módszert alkalmaztuk és sikeresen kapcsoltuk a 2-O-acetil-4,6-O-(S)-(1-metoxikarbonil-etilidén)-3-O-metil-D-glükopi-ranozil-triklór-acetimidát donort a p-Nitrofenil-endo-3,4-O-benzilidén-6-dezoxi-2-O-(2,4-di-O-benzil-a-L-ramnopirano-zil)-a-L-talopiranozid akceptorral, nyerve célvegyületünket, a p-Nitrofenil-2-O-{3-O-[2-O-acetil-4,6-O-(S)-(1-metoxikar-bonil-etilidén)-3-O-metil-b-D-glükopiranozil]-2,4-di-O-ben-zil-a-L-ramnopiranozil}-endo-3,4-O-benzilidén-6-dezoxi-a-L-talopiranozid triszacharidot.
Synthesis of oligosaccharides of mycobacterial origin
Ph.D. thesis, Lajos Kossuth University, Debrecen
1996
This dissertation deals with the chemical synthesis of natural oligosaccharides of Mycobacterial origin.
The structure of the haptenic trisaccharide of the Mycobactereium avium serovar 14 is shown on Figure A.
The nonreducing terminus of this pentasaccharide contains a branched-chain sugar with unique structure called N-formylkansosamine (4,6-dideoxy-4-formamido-3-C-meth-yl-2-O-methyl-L-mannose). The penultimate unit is a substituted D-rhamnose which occurs rarely in natural oligosaccharides (Figure A).
For the synthesis of the N-formylkansosaminewe used the strategy developed by Klemer et. al. for C-methylation Using this we were able to prepare methyl-6-deoxy-2,3-O-isopropylidene-3-C-methyl-a-L-lyxo-hexopyranoside-4-ulose in excellent yield. The reduction of the oxime of this ulose with LiAlH4 then gave a mixture of amines with 4,6-dideoxy-manno- and 4,6-dideoxy-talo- configuration. These were purified with chromatography and formylated using acetic-formic anhydride. After the above crucial synthetic steps and few protection - deprotection manipulations the 3-O-benzyl-4-N-benzylformamido-4,6-dideoxi-3-C-methyl-2-O-methyl-a-L-mannopyranosyl trichloroacetimidate N-formylkansosaminyl donor was prepared. We synthesized also the phenylthio N-formylkansosaminyl donor using the above strategy.
For the preparation of the p-nitrophenyl-4-O-benzyl-6-deoxy-2-O-methyl-a-D-mannopyranoside acceptor the p-nitrophenyl-a-D-mannopyranoside was used as starting material. The selective reduction of the 6-OTs group of p-nitrophenyl-2,3-O-isopropylidene-6-O-p-toluenesulfonyl-a-D-mannopyranoside was unsuccesful, thus the 6-OTs group was transformed to 6-I functionality. Treatment of the 6-deoxy-6-iodo derivative with LiAlH4 then resulted in the desired 6-deoxy compound. After benzylation and acidic hydrolysis of the isopropylidene group, the resulting diol was selectively O-methylated at position 2 using phase-transfer catalysis to yield the p-nitrophenyl-4-O-benzyl-6-deoxy-2-O-methyl-a-D-mannopyranoside as a suitable glycosyl acceptor for the synthesis of the terminal disaccharide.
For the synthesis of 3-O-benzyl-4-N-benzylformamido-4,6-dideoxi-3-C-methyl-2-O-methyl-a-L-mannopyranosyl-(1®3)-4-O-benzyl-6-deoxi-2-O-methyl-a-D-mannopyra-nosyl-(1®OPNP) and the pentasaccharide (the latter with 1+4 glycosylation strategy) the phenyl-1-thio kansosaminyl donor was unsuitable. The trichloroacetimidate donor and the p-nitrophenyl-4-O-benzyl-6-deoxy-2-O-methyl-a-D-mannopyranoside acceptor gave the terminal disaccharide in a yield of 35 %, but again, the 1+4 strategy was unsuccesful with the trichloroacetimidate donor. Thus, for the preparation of the pentasaccharide, we synthesized the 3-O-benzyl-4-N-benzylformamido-4,6-dideoxi-3-C-methyl-2-O-methyl-a-L-mannopyranosyl-(1®3)-4-O-benzyl-6-deoxi-2-O-methyl-a-D-mannopyranosyl-(1®SEt) disaccharide donor, and with this donor and the 4-O-benzyl-2-O-methyl-a-L-fucopyranosyl-(1®3)-2,4-di-O-benzyl-6-deoxi-a-L-man-nopyranosyl-(1®2)-3,4-O-endo-benzylidene-a-L-talopyra-nosyl-(1®OPNP) acceptor the 2 + 3 strategy for the synthesis of the target pentasaccharide was succesful.
The structure of the haptenic trisaccharide of the Mycobactereium avium serovar 8 is shown on Figure B.
The 4,6-O-(S)-1-carboxyethylidene-3-O-methyl-b-D-glucopyranosyl unit was prepared starting from benzyl 2-O-benzyl-3-O-methyl-b-D-glucopyranoside using methyl 2,2-di(ethylthio) propionate as the pyruvylating agent and SO2Cl2-TfOH as catalyst. The diastereoisomers formed were isolated by column chromatography and the S isomer was then hydrogenated, acetylated, and the anomeric acetyl group was removed with hydrazine-acetate to give 2-O-acetyl-4,6-O-(S)-(1-methoxycarbonylethylidene)-3-O-methyl-D-glucopyranose. This was then transformed into the trichloroacetimidate donor.
The earlier reported synthesis of the suitably protected crystalline 2,4-di-O-benzyl-6-deoxi-a-L-mannopyranosyl-(1®2)-3,4-O-endo-benzylidene-a-L-talopyranosyl-(1®OPNP) disaccharide glycosyl acceptor was slightly modified in that a new rhamnosyl donor, the ethyl 3-O-acetyl-2,4-di-O-benzyl-1-thio-a-L-rhamnopyranoside was applied. For the preparation of this new donor we utilized our earlier observation that phase-transfer alkylation of thioglycosides at position 2 proceeds more easily and with better selectivity than the alkylation of O-glycosides. In the present case, ethyl 4-O-benzyl-1-thio-a-L-rhamnopyra-noside was benzylated under phase-transfer catalysis conditions to produce ethyl 2,4-di-O-benzyl-1-thio-a-L-rhamnopyranoside in a yield of 64 %. Conventional acetylation then gave the proper rhamnosyl donor. Glycosylation of p-nitrophenyl endo-3,4-O-benzylidene-a-L-talopyranoside with the above donor and subsequent deacetylation of O-3 of the prepared disaccharide then yielded the crystalline disaccharide acceptor.
Glycosylation of the core disaccharide acceptor with 2-O-acetyl-4,6-O-(S)-(1-methoxycarbonylethylidene)-3-O-methyl-D-glucopyranosyl trichloroacetimidate in the presence of trimethylsilyl trifluoromethanesulfonate promoter at -50 °C furnished the fully protected trisaccharide (42 %). The b configuration of the newly formed glycosidic linkage was proved by careful analysis of the 1H and 13C NMR spectra, which also revealed that, fortunately, no isomerization at the acetalic carbon occured during the glycosylation.
Publications
1. I. Bajza and A. Lipták, Carbohydr. Res., 205, 435-439, (1990).
2. I. Bajza, J. Kerékgyártó, J. Hajkó, L. Szilágyi, and A. Lipták, Carbohydr. Res., 253, 111-120, (1993).
3. A. Lipták, J. Kerékgyártó, and I. Bajza, Proceedings of the Second Korea-Hungary Symposium on Organic Chemistry, Seoul, November 4-5, pp. 25-42, (1993).
4. A. Lipták, A. Borbás, and I. Bajza, Medicinal Res. Reviews, 14, 307-352, (1994).
5. Bajza I., Borbás A., Hajkó J., Lagas, R., Szabovik G., Varga Zs., Lipták A., Magy. Kém. Lapja, 51, 000, (1996).
| Vissza a tartalomjegyzékhez Back to Contents |
http://www.kfki.hu/chemonet/ http://www.ch.bme.hu/chemonet/ |